A typical human cell contains billions of protein molecules ranging in abundance from low copy numbers to millions of copies. This vast scale of the proteome poses major challenges for any method aiming for comprehensive protein analysis. It is a fundamental reason why proteomics is challenging.
Despite the challenges, our ability to quantify proteins from complex samples has grown remarkably fast, with about 1,250-fold increase in the throughput of mass spec proteomics from 2001 to 2021. Here, I will focus on recent results that exemplify this growth, namely quantifying over 100,000 precursors from single cells analyzed on a 15 min active gradient. Let’s put these results in context.
They reminded me of a wonderful big-picture JPR article by Annette et al., 2021 describing the detection of over 100,000 peptide-like features within 60 min of active gradient. The authors emphasized the challenges of determining their sequences by data dependent acquisition (DDA). As a result, only a small fraction of these peptide-like features could be matched to peptide sequences, Fig. 1. The limitations of DDA are further compounded for sensitive analysis of single cells by the need to accumulate precursors for longer periods of time before having enough ion copies for robust MS spectra.

This challenge was overcome by a paradigm shift in data acquisition and interpretation: A transition from isolating and fragmenting one precursor at a time (by DDA) to isolating and fragmenting many precursors at a time (by DIA, data independent acquisition). By multiplexing DIA, Derks et al., 2021 were able to identify 235,932 precursors in a single plexDIA run within 60 min of active gradient, Fig. 1.
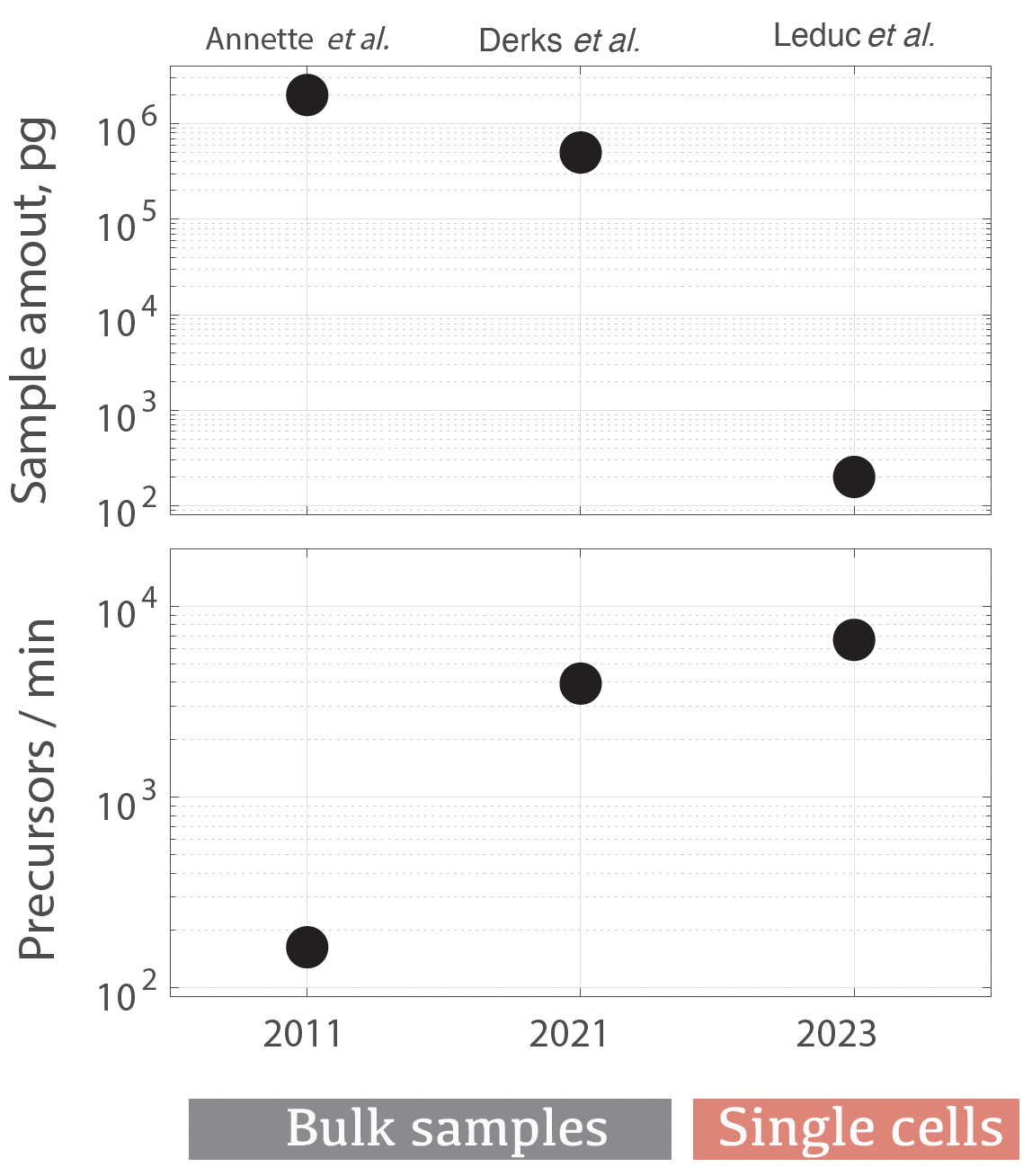
Figure 1 | Sensitivity (input sample amount) and throughput (# of precursors identified per minute of active gradient) in 3 experiments by Annette et al., 2021, Derks et al., 2021 / Derks et al., 2022, and Leduc et al., 2023.
Further advances in MS instrumentation (timsTOF Ultra), sample preparation and data analysis enabled the identification of about 100,000 precursors in just 15 min of analysis of single cells. What was not possible to do in 2011 with bulk samples, now can be performed with single cells in just a small fraction of the time, Fig. 1.
Many factors combine synergistically to contribute to this remarkable progress. These factors have much space to grow, and thus we expect to see sustained progress in MS-based proteomics.
In my opinion, this technological progress will unfold huge opportunities for biomedical research.
